BALSAMIC Variant Calling Algorithms¶
In BALSAMIC, various bioinfo tools are integrated for reporting somatic and germline variants. Also, the choice of these tools differs between the type of analysis, for eg: Target Genome Analysis (TGA) or Whole Genome Sequencing (WGS). Various filters (Pre-call filtering and Post-call filtering) are applied at different levels to report high-confidence variant calls.
Pre-call filtering is where the variant-calling tool decides not to call a variant line to the VCF file, if the default filters did not pass the criteria. The set of default filters differs between the various variant-calling algorithms.
To know more about the pre-call filters used by the variant callers, please have a look at the VCF header of the particular variant-calling results. For example:
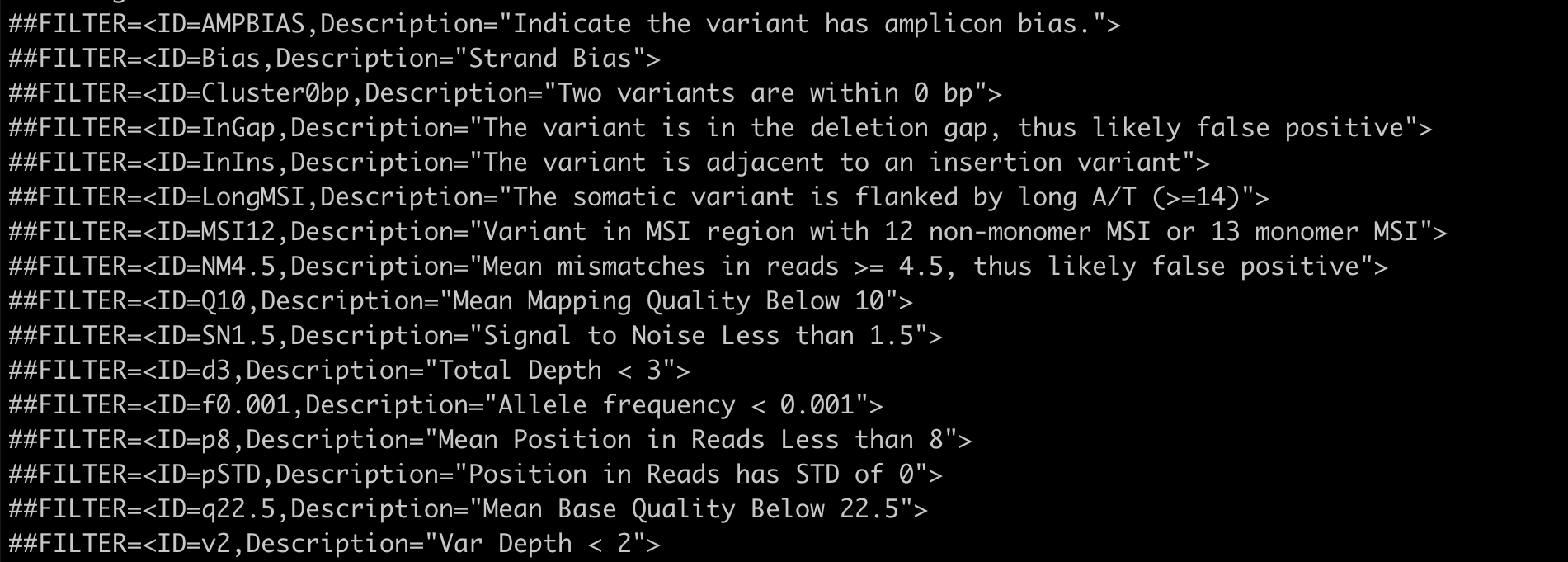
Pre-call filters applied by the Vardict variant-caller is listed out in the VCF header¶
In the VCF file, FILTER status is PASS if this position has passed all filters, i.e., a call is made at this position. Otherwise, if the site has not passed all filters, a semicolon-separated list of codes for filters that fail. e.g., p8;pSTD might indicate that at this site, the mean position in reads is less than 8 and position in reads has a standard deviation of 0.
Important
In BALSAMIC, this VCF file is named as *.all.vcf.gz (eg: SNV.somatic.<CASE_ID>.vardict.all.vcf.gz)
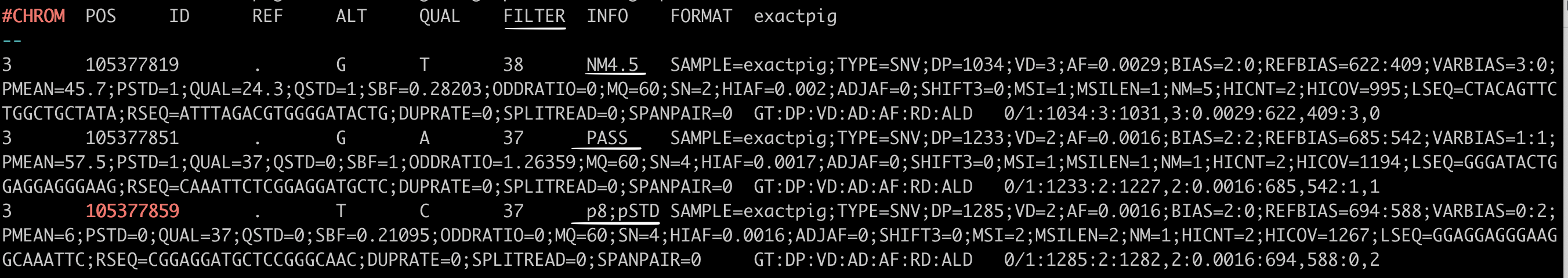
Vardict Variant calls with different ‘FILTER’ status underlined in white line (NM4.5, PASS, p8;pSTD)¶
Post-call filtering is where a variant is further filtered with criteria such as quality, depth, VAF etc with more stringent thresholds.
For Post-call filtering, in BALSAMIC we have applied various filtering criteria (Vardict_filtering, TNscope filtering (Tumor_normal) ) depending on the analysis-type (TGS/WGS) and sample-type(tumor-only/tumor-normal).
Important
In BALSAMIC, this VCF file is named as *.all.filtered.pass.vcf.gz (eg: SNV.somatic.<CASE_ID>.vardict.all.filtered.pass.vcf.gz)
Targeted Genome Analysis¶
Somatic Callers for reporting SNVs/INDELS¶
Vardict¶
Vardict is a sensitive variant caller used for both tumor-only and tumor-normal variant calling. The results of Vardict variant calling are further post-filtered based on several criteria (Vardict_filtering) to retrieve high-confidence variant calls. These high-confidence variant calls are the final list of variants uploaded to Scout or available in the delivered VCF file in Caesar.
Vardict_filtering¶
Following are the set of criterias applied for filtering vardict results. Applies for both tumor-normal and tumor-only samples
Mean Mapping Quality (MQ): Refers to the root mean square (RMS) mapping quality of all the reads spanning the given variant site.
MQ >= 40
Total Depth (DP): Refers to the overall read depth supporting the called variant.
DP >= 100
Variant depth (VD): Total reads supporting the ALT allele
VD >= 5
Allelic Frequency (AF): Fraction of the reads supporting the alternate allele
Minimum AF >= 0.007
Maximum AF < 1
Attention
BALSAMIC <= v8.2.7 uses minimum AF 1% (0.01). From Balsamic v8.2.8, minimum VAF is changed to 0.7% (0.007)
GNOMADAF_POPMAX: Maximum Allele Frequency across populations
GNOMADAF_popmax <= 0.005 (or) GNOMADAF_popmax == "."
Important
Additionally, for tumor-normal cases; the variant is excluded if it marked as ‘germline’ in the STATUS column of vcf file.
Whole Genome Sequencing (WGS)¶
Sentieon’s TNscope¶
BALSAMIC utilizes TNscope algorithm for the variant calling of somatic SNV/INDELS in WGS samples. The TNscope algorithm performs the somatic variant calling on the tumor-normal or the tumor-only samples, using a Haplotyper algorithm.
TNscope filtering (Tumor_normal)¶
Total Depth (DP): Refers to the overall read depth from all target samples supporting the variant call
DP(tumor) >= 10 || DP(normal) >= 10
Allelic Depth (AD): Total reads supporting the ALT allele in tumor sample
AD(tumor) >= 3
Allelic Frequency (AF): Fraction of the reads supporting the alternate allele
Minimum AF(tumor) >= 0.05
Maximum AF(tumor) < 1
GNOMADAF_POPMAX: Maximum Allele Frequency across populations
GNOMADAF_popmax <= 0.001 (or) GNOMADAF_popmax == "."
TNscope filtering (tumor_only)¶
Total Depth (DP): Refers to the overall read depth supporting the variant call
DP(tumor) >= 10
Allelic Depth (AD): Total reads supporting the ALT allele in tumor sample
AD(tumor) > 3
Allelic Frequency (AF): Fraction of the reads supporting the alternate allele
Minimum AF(tumor) > 0.05
Maximum AF(tumor) < 1
GNOMADAF_POPMAX: Maximum Allele Frequency across populations
GNOMADAF_popmax <= 0.001 (or) GNOMADAF_popmax == "."
Normalized base quality scores: The sum of base quality scores for each allele (QSS) is divided by the allelic depth of alt and ref alleles (AD)
SUM(QSS)/SUM(AD) >= 20
Read Counts: Count of reads in a given (F1R2, F2R1) pair orientation supporting the alternate allele and reference alleles
ALT_F1R2 > 0, ALT_F2R1 > 0
REF_F1R2 > 0, REF_F2R1 > 0
SOR: Symmetric Odds Ratio of 2x2 contingency table to detect strand bias
SOR < 3
Target Genome Analysis with UMI’s into account¶
Sentieon’s TNscope¶
UMI workflow performs the variant calling of SNVs/INDELS using the TNscope algorithm from UMI consensus-called reads. The following filter applies for both tumor-normal and tumor-only samples.
Pre-call Filters
minreads: Filtering of consensus called reads based on the minimum reads supporting each UMI tag group
minreads = 3,1,1
Which means that at least 3 UMI tag groups should be ideally considered from both DNA strands, where a minimum of atleast 1 UMI tag group should exist in each of the single-stranded consensus reads.
min_init_tumor_lod : Log odds is the likelihood that the candidate mutation is real over the likelihood that the candidate mutation is a sequencing error before any read-based filters are applied. minimum log odds for the candidate selection. TNscope default: 4
min_init_tumor_lod = 0.5
min_tumor_lod : minimum log odds in the final call of variants. TNscope default: 6.3
min_tumor_lod = 4.0
Post-call Filters
GNOMADAF_POPMAX: Maximum Allele Frequency across populations
GNOMADAF_popmax <= 0.001 (or) GNOMADAF_popmax == "."