Frequently Asked Questions (FAQs)#
BALSAMIC#
UMIworkflow#
What are UMIs
Unique Molecular Identifiers (UMIs) are short random nucleotide sequences (3-20 bases) that are ligated to the ends of DNA fragments prior to sequencing to greatly reduce the impact of PCR duplicates and sequencing errors on the variant calling process.
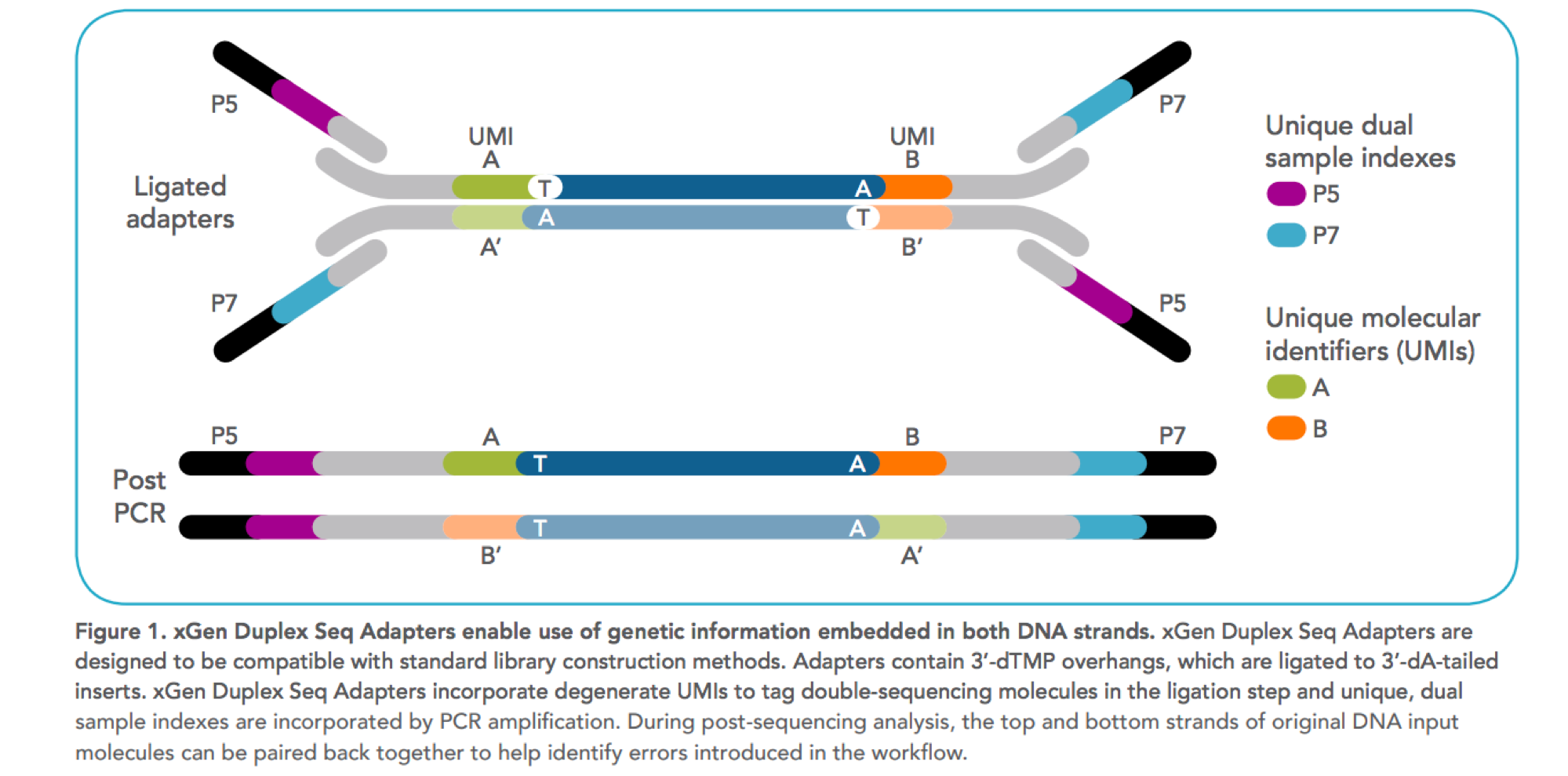
Figure1: Design of UMI adapters in the library preparation. Ref#
How is the UMIworkflow implemented
CG’s UMIworkflow is implemented using the commercial software Sentieon. The Sentieon tools provide functionality for extracting UMI tags from fastq reads and performing barcode-aware consensus generation. The workflow is as described:
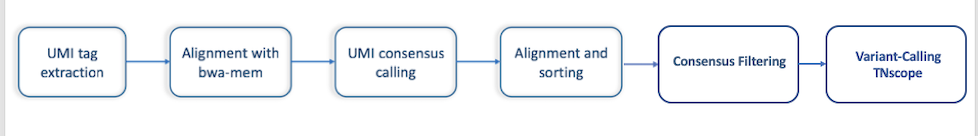
Figure2: UMI workflow steps.#
How is the UMI structure defined
Our pair-end sequencing read length is about 151 bp and the UMI structure is defined as`3M2S146T, 3M2S146T` where 3M represents 3 UMI bases, 2S represents 2 skipped bases, 146T represents 146 bases in the read.
Are there any differences in the UMI read extraction if the read structure is defined as `3M2S146T, 3M2S146T` or `3M2S+T, 3M2S+T`?
In theory, this should be the same if the read length is always 151bp. But the recommendation is to use 3M2S+T, 3M2S+T so that UMIworkflow can handle any unexpected input data.
How does the `umi extract` tool handle sequencing adapters? Do the input reads always need to be adapter removed fastq reads
The presence of 5’ adapter sequences can cause issues for the Sentieon umi extract tool, as the extract tool will not correctly identify the UMI sequence. If 5’ adapter contamination is found in the data, before processing with the umi extract tool, these adapter sequences needed to be removed with a third-party trimming tool. 3’ adapter contamination is much more common and can occur when the insert size is shorter than the sequence read length. The Sentieon umi consensus tool will correctly identify and handle 3’ adapter/barcode contamination during consensus read creation.
How does Sentieon `umi consensus` tool handles paired-end reads
The umi consensus tool will merge overlapping read pairs when it can, but it is not possible for reads with an insert size greater than 2x the read length as there is some unknown intervening sequence. In this case, umi consensus will output a consensus read pair where each consensus read in the pair is constructed separately, while other reads in the dataset are collapsed/merged to single-end reads.
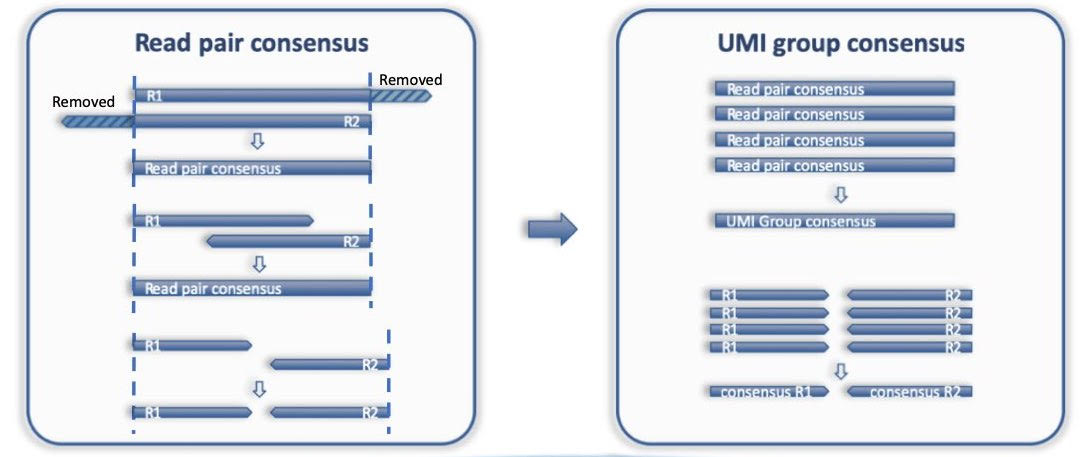
Figure3: Figure taken from Sentieon document.#
Purpose of consensus-filtering step in the UMIworkflow
Mainly to reduce the calling of false-positive variants. Consensus filtering is based on the setting of minimum raw reads (MinR) supporting each UMI group. By default, MinR is set as 3,1,1, meaning that the minimum number of raw reads in both strands should be greater than 1 and the sum of both strands is greater than 3. The default 3,1,1 is a good starting point at lower coverages. This setting can be further adjusted accordingly at higher coverages or if finding false-positive calls due to consensus reads with little read support.
How is the performance of other variant callers for analysing UMI datasets UMI workflow is validated with two datasets (SeraCare and HapMap). The Vardict failed to call the true reference variants while the TNscope performed better. A more detailed analysis is summarized here
We are still investigating other UMI-aware variant callers and maybe in the future, if something works better, additional varcallers will be added to the UMIworkflow.
Git Related Questions#
How to make a new release of balsamic
Our release model looks like in the figure below:
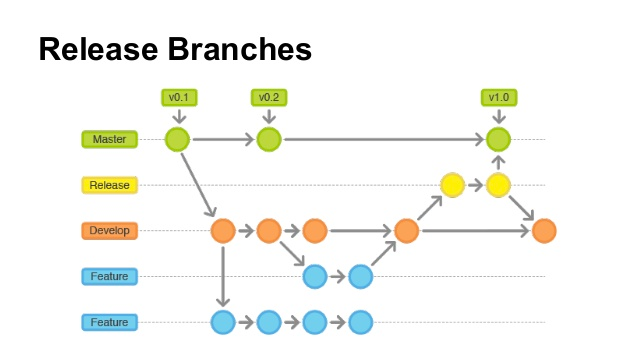
If changes are hotfixes/patches, make a release from the master branch. Otherwise, make a release from the develop branch.
Release from develop:
Switch to the master branch git checkout master.
Pull the latest changes git pull.
Switch to the develop branch: git checkout develop.
Merge the latest changes from master into develop: git merge master.
Resolve any conflicts, commit the changes, and push them to the develop branch.
Create a new release branch: git checkout -B release_vX.X.X. Adhere strictly to the naming convention for compatibility with container publishing GitHub Actions. Failure to do so may result in the unavailability of containers for download in this particular version.
Update the version number X.X.X in the CHANGELOG.rst.
Open a pull request for the release branch, and after obtaining approval, merge it into master.
Move back to master branch: git checkout master.
Fetch the latest changes: git pull.
Use bumpversion to increment the version (choose major, minor, or patch): bumpversion –verbose [major/minor/patch].
Push the version increment changes: git push.
Push the tags for the new version: git push –tags.
Merge the changes into the develop branch (git checkout develop && git merge master).
Use the release branch to verify/validate the new version.
Once the verification/validation process is successfully completed and approved, proceed with the deployment to production.
Note
Never force rebase commits into master or develop.
For pull requests to master, opt for Create a merge commit to capture full commit history.
For pull requests to develop, use Squash and merge to combine commit messages.