Calling and filtering variants#
In BALSAMIC, various bioinfo tools are integrated for reporting somatic and germline variants summarized in the table below. The choice of these tools differs between the type of analysis; Whole Genome Sequencing (WGS), or Target Genome Analysis (TGA) and Target Genome Analysis (TGA) with UMI 3,1,1 filtering activated.
Variant caller |
Sequencing type |
Analysis type |
Somatic/Germline |
Variant type |
|---|---|---|---|---|
DNAscope |
TGA, WGS |
tumor-normal, tumor-only |
germline |
SNV, InDel |
TNscope |
WGS, TGA, TGA with UMI 3,1,1 filtering applied |
tumor-normal, tumor-only |
somatic |
SNV, InDel |
VarDict |
TGA |
tumor-normal, tumor-only |
somatic |
SNV, InDel |
Various filters (Pre-call and Post-call filtering) are applied at different levels to report high-confidence variant calls.
Pre-call filtering is where the variant-calling tool decides not to add a variant to the VCF file if the default filters of the variant-caller did not pass the filter criteria. The set of default filters differs between the various variant-calling algorithms.
To know more about the pre-call filters and detailed arguments used by the variant callers, please have a look at the VCF header of the particular variant-calling results. For example:
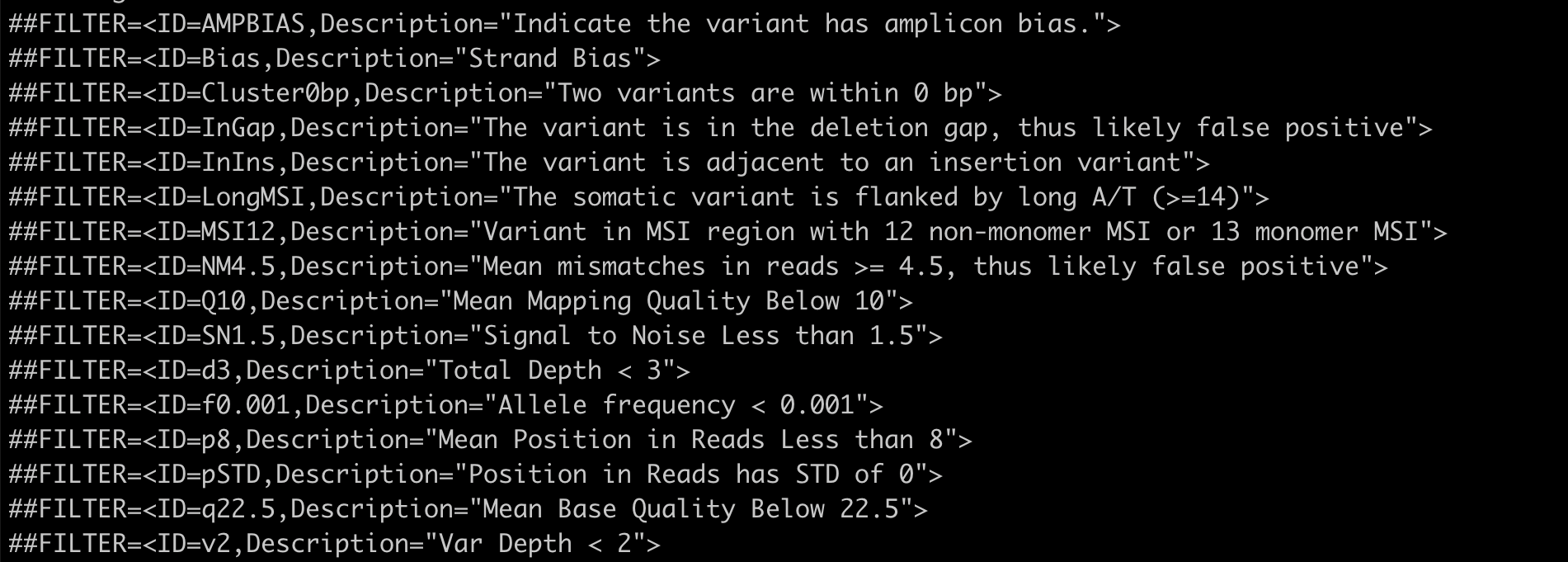
Pre-call filters applied by the TNscope variant-caller is listed in the VCF header.#
In the VCF file, the FILTER status is PASS if this position has passed all filters, i.e., a call is made at this position. Contrary, if the site has not passed any of the filters, a semicolon-separated list of those failed filter(s) will be appended to the FILTER column instead of PASS. E.g., t_lod_fstar;alt_allele_in_normal might indicate that at this site there is little support that the alternative bases constitute a true somatic variant, and there is also evidence of those same bases in the normal sample.
Note: In BALSAMIC, this VCF file is often referred to as the “raw” VCF because it is the most unfiltered VCF produced, and is named `SNV.somatic.<CASE_ID>.tnscope.vcf.gz`
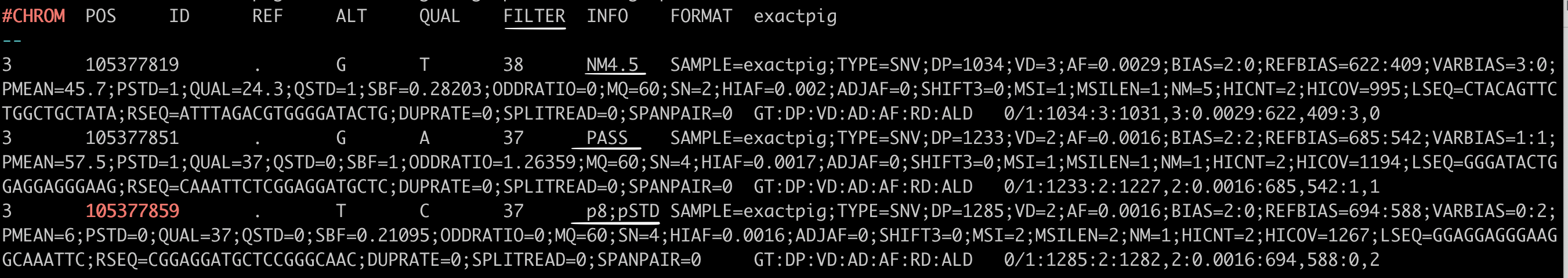
TNscope Variant calls with different ‘FILTER’ status underlined in red line (PASS, t_lod_fstar, alt_allele_in_normal)#
Post-call quality filtering is where a variant is further filtered based on quality, depth, VAF, etc., with more stringent thresholds.
For Post-call filtering, in BALSAMIC we have applied various filtering criteria depending on the analysis-type (TGS/WGS) and sample-type (tumor-only/tumor-normal), and only variants with either PASS or triallelic_site are kept.
Note: In BALSAMIC, this VCF file is named as SNV.somatic.<CASE_ID>.research.tnscope.vcf.gz and is not delivered
Post-call variant-database filtering is where a variant is further filtered based on their presence in certain variant-databases such as Gnomad and local variant databases built with LoqusDB.
This is a two step process where variants are first filtered based on existing above a specified frequency in public available databases, and then based on local databases of previously observed variants.
At each step only variants with filters PASS and triallelic_site are kept and delivered as a final list of variants to the customer either via Scout or Caesar
Note: In BALSAMIC, the VCF file filtered only on public available databases is named as `*.research.filtered.pass.vcf.gz` (eg: for WGS `SNV.somatic.<CASE_ID>.tnscope.research.filtered.pass.vcf.gz`) In BALSAMIC, the VCF file filtered on both public and locally available databases is named as `*.clinical.filtered.pass.vcf.gz` (eg: for TGA `SNV.somatic.<CASE_ID>.merged.<research/clinical>.filtered.pass.vcf.gz`)
VCF file name |
Description |
Delivered to the customer |
|---|---|---|
.vcf.gz |
Unannotated raw VCF file with pre-call filters included in the STATUS column |
Yes (Caesar) |
.research.filtered.pass.vcf.gz |
Annotated VCF file with quality and population based filters applied. |
Yes (Caesar) |
.clinical.filtered.pass.vcf.gz |
Annotated VCF file with quality, population and local database filters applied. |
Yes (Caesar and Scout) |
Soft-filters
These soft-filters are never applied, meaning variants with these filters set will be present in the final filtered VCF.
Soft-filters |
Sequencing type |
Analysis type |
Somatic/Germline |
Description |
|---|---|---|---|---|
triallelic_site |
TGA, WGS |
tumor-normal, tumor-only |
somatic |
internal TNscope filter due to more than 1 alt allele |
germline_risk |
TGA, TGA - UMI 3,1,1 |
tumor-normal |
somatic |
internal TNscope filter implying risk of germline variant |
in_normal |
TGA, TGA - UMI 3,1,1 |
tumor-normal |
somatic |
Custom bcftools filter based on high relative normal vs tumor alt allele fractions |
HighOccurrenceFrq |
TGA, TGA - UMI 3,1,1 (if optional panel-observation-loqusdb argument is supplied) |
tumor-normal |
somatic |
If presence of variant is high in panel-based observation database |
Targeted Genome Analysis#
Regarding matched normal analyses#
Since Balsamic v17.0.0 the option –soft-filter-normal was added and automatically applied for all Targeted Genome Analyses with a matched normal.
This option disables hard-filtering on the matched normal specific filters; germline_risk from TNscope and the in_normal custom bcftools filter mentioned below under Relative tumor AF in normal.
These matched normal soft-filters can optionally be applied out in Scout to revert to the original hard-filter behaviour.
Somatic Callers for reporting SNVs/INDELS#
For SNV/InDel calling in the TGA analyses of balsamic both VarDict and TNscope are used. Lists of variants are produced from both tools, which are then normalised and quality filtered before being merged with a custom made python script which can be found in BALSAMIC/assets/scripts/merge_snv_variantcallers.py.
The requirement for merging variants with this script is a perfect match of; CHROM, POS, REF and ALT fields.
The INFO fields from both VCFs are merged entirely, and when the same field exists in both variants it is converted to a comma-separated list. An exception to this behaviour is the AF and DP fields for which the single values are maintained (from the first VCF in the positional argument), and new fields called AF_LIST and DP_LIST are created which contains a list of values from both callers.
Calling and quality filtration#
This section focuses on the calling and quality filtration done on VarDict and TNscope variant calls.
Vardict#
Vardict is a sensitive variant caller used for both tumor-only and tumor-normal variant calling.
There are two slightly different post-processing filters activated depending on if the sample is an exome or a smaller panel as these tend to have very different sequencing depths.
Vardict_filtering#
Following is the set of criteria applied for filtering vardict results. It is used for both tumor-normal and tumor-only samples.
Post-call Quality Filters
Mean Mapping Quality (MQ): Refers to the root mean square (RMS) mapping quality of all the reads spanning the given variant site.
MQ >= 30
Total Depth (DP): Refers to the overall read depth supporting the called variant.
DP >= 50
Variant depth (VD): Total reads supporting the ALT allele
VD >= 5
Allelic Frequency (AF): Fraction of the reads supporting the alternate allele
Minimum AF >= 0.005
Post-call Quality Filters for specific for exomes
Total Depth (DP): Refers to the overall read depth supporting the called variant.
DP >= 20
Attention: BALSAMIC <= v8.2.7 uses minimum AF 1% (0.01). From Balsamic v8.2.8, minimum VAF is changed to 0.7% (0.007). From v16.0.0 minimum VAF is changed to 0.5% (0.005).
Specific for VarDict normal matched analyses
Relative tumor AF in normal: Allows for maximum Tumor-In-Normal-Contamination of 30%.
marks variant with soft-filter `in_normal` variant if: AF(normal) / AF(tumor) > 0.3
Sentieon’s TNscope#
The TNscope algorithm performs the somatic variant calling on the tumor-normal or the tumor-only samples.
TNscope filtering#
Pre-call Filters
min_init_tumor_lod: Initial Log odds for the that the variant exists in the tumor.
min_init_tumor_lod = 0.5
min_tumor_lod: Minimum log odds in the final call of variant in the tumor.
min_tumor_lod = 4
min_init_normal_lod: Initial Log odds for the that the variant exists in the normal.
min_init_normal_lod = 0.5
min_normal_lod: Minimum log odds in the final call of variant in the normal.
min_normal_lod = 2.2
min_dbnp_normal_lod: Minimum normalLOD at dbSNP site.
min_dbnp_normal_lod = 5.5
max_error_per_read: Maximum number of differences to reference per read.
max_error_per_read = 5
min_base_qual: Minimal base quality to consider in calling
min_base_qual = 15
min_tumor_allele_frac: Set the minimum tumor AF to be considered as potential variant site.
min_tumor_allele_frac = 0.0005
interval_padding: Adding an extra 100bp to each end of the target region in the bed file before variant calling.
interval_padding = 100
Post-call Quality Filters
Total Depth (DP): Refers to the overall read depth supporting the called variant.
DP >= 50
Variant depth (VD): Total reads supporting the ALT allele
VD >= 5
Allelic Frequency (AF): Fraction of the reads supporting the alternate allele
Minimum AF >= 0.005
RPA: Number of times tandem repeat unit is repeated, for each allele (including reference)
RPA < 12
Post-call Quality Filters for specific for exomes
Total Depth (DP): Refers to the overall read depth supporting the called variant.
DP >= 20
Specific for TNscope tumor only analyses
Average base quality score
SUM(QSS)/SUM(AD) >= 20
SOR: Symmetric Odds Ratio of 2x2 contingency table to detect strand bias
SOR < 2.7
Note: Additionally, variants labeled with triallelic site filter are not filtered out
Specific for TNscope normal matched analyses
SOR: Symmetric Odds Ratio of 2x2 contingency table to detect strand bias
SOR < 3
alt_allele_in_normal: Default filter set by TNscope was considered too stringent in filtering tumor in normal and is removed.
bcftools annotate -x FILTER/alt_allele_in_normal
Relative tumor AF in normal: Allows for maximum Tumor-In-Normal-Contamination of 30%.
marks variant with soft-filter `in_normal` variant if: AF(normal) / AF(tumor) > 0.3
Post-processing of TNscope variants
After quality-filtering TNscope variants and before merging with VarDict variants the phased SNVs and InDels from TNscope are merged together to MNVs using a slightly modified script from Sentieon-scripts which can be found in BALSAMIC/assets/scripts/merge_mnp.py
This was done to avoid multiple representations of the same variant as VarDict already outputs these types of variants as MNVs, and because VEP isn’t coded to handle phased SNVs in the interpretation of protein effect.
In the merging of phased SNVs to MNV we need to handle how to consolidate information from multiple variants into a single metric, and importantly also for the FILTER column.
An example is a MNV created by merging a phased germline SNV with a somatic SNV. This has been solved as follows:
MNV_CONFLICTING_FILTERS: Is a filter given to MNVs with constituent variants with different filters (such as in_normal and PASS)
However, as we may have multiple filters which means essentially the same thing, such as germline_risk and in_normal, MNVs created from the merging of variants with only those filters aren’t actually conflicting.
Therefore the logic for setting MNV_CONFLICTING_FILTERS has been made a bit more complex, and in summary there are 3 possible outcomes for filters when merging SNVs/InDels into MNVs:
Single filter such as PASS, when all constituting variants all have the same filter and no other.
Multiple filters, such as in_normal,germline_risk, when all constituting variants have at least 1 of the matched normal filters.
MNV_CONFLICTING_FILTERS when the merged variants have conflicting filters, and they don’t all contain matched normal filters.
Note
In addition to this a few more fields are added to the INFO field of the created MNVs containing comma-separated lists of AD, AF, and FILTER from its constituting variants.
Post-call Observation database Filters#
Note
Since BALSAMIC <= v18.0.0 Post-call database filters are initially applied as soft-filters and can be whitelisted in a subsequent step, whereupon they will be PASS
This section contains post call and quality filtrations, on the TNscope and VarDict merged VCF.
GNOMADAF_POPMAX: Maximum Allele Frequency across populations
GNOMADAF_popmax <= 0.005 (or) GNOMADAF_popmax == "."
SWEGENAF: SweGen Allele Frequency
SWEGENAF <= 0.01 (or) SWEGENAF == "."
Frq: Frequency of observation of the variants from normal Clinical samples
Frq <= 0.01 (or) Frq == "."
ArtefactFrq: Frequency of observation of the variants from normal WGS samples merged to ~1200X coverage
ArtefactFrq <= 0.1 (or) ArtefactFrq == "."
This above corresponds to at least 4 observations in a database of 29 cases of merged WGS samples.
Cancer_Somatic_Panel_Frq: Frequency of observation of the variants from historical cases analysed with the same gene-panel
marks variant with soft-filter `HighOccurrenceFrq` variant unless: Cancer_Somatic_Panel_Frq <= 0.3 (or) Cancer_Somatic_Panel_Frq == "."
Target Genome Analysis with UMI’s into account#
This section contains specific filters and settings for the balsamic-umi workflow, which filters on UMI group size (default 3,1,1) before variant-calling.
Calling and quality filtration#
This section focuses on calling and quality filters.
Sentieon’s TNscope#
UMI workflow performs the variant calling of SNVs/INDELS using the TNscope algorithm from UMI consensus-called reads. The following filter applies for both tumor-normal and tumor-only samples.
Pre-call Filters
minreads: Filtering of consensus called reads based on the minimum reads supporting each UMI tag group
minreads = 3,1,1
It means that at least 3 read-pairs need to support the UMI-group (based on the UMI-tag and the aligned genomic positions), and with at least 1 read-pair from each strand (F1R2 and F2R1). NOTE: This filtering is performed on the bamfile before variant calling.
min_init_tumor_lod: Initial Log odds for the that the variant exists in the tumor.
min_init_tumor_lod = 0.5
min_tumor_lod: Minimum log odds in the final call of variant in the tumor.
min_tumor_lod = 4
min_init_normal_lod: Initial Log odds for the that the variant exists in the normal.
min_init_normal_lod = 0.5
min_normal_lod: Minimum log odds in the final call of variant in the normal.
min_normal_lod = 2.2
min_dbnp_normal_lod: Minimum normalLOD at dbSNP site.
min_dbnp_normal_lod = 5.5
max_error_per_read: Maximum number of differences to reference per read.
max_error_per_read = 5
min_base_qual: Minimal base quality to consider in calling
- ::
min_base_qual = 15
min_tumor_allele_frac: Set the minimum tumor AF to be considered as potential variant site.
min_tumor_allele_frac = 0.0005
interval_padding: Adding an extra 100bp to each end of the target region in the bed file before variant calling.
interval_padding = 100
Post-call Quality Filters
alt_allele_in_normal: Default filter set by TNscope was considered too stringent in filtering tumor in normal and is removed.
bcftools annotate -x FILTER/alt_allele_in_normal
Relative tumor AF in normal: Allows for maximum Tumor-In-Normal-Contamination of 30%.
marks variant with soft-filter `in_normal` variant if: AF(normal) / AF(tumor) > 0.3
Post-call Observation database Filters#
Note
Since BALSAMIC <= v18.0.0 Post-call database filters are initially applied as soft-filters and can be whitelisted in a subsequent step, whereupon they will be PASS
This section contains population database frequency filters.
GNOMADAF_POPMAX: Maximum Allele Frequency across populations
GNOMADAF_popmax <= 0.02 (or) GNOMADAF_popmax == "."
SWEGENAF: SweGen Allele Frequency
SWEGENAF <= 0.01 (or) SWEGENAF == "."
Frq: Frequency of observation of the variants from normal Clinical samples
Frq <= 0.01 (or) Frq == "."
Cancer_Somatic_Panel_Frq: Frequency of observation of the variants from historical cases analysed with the same gene-panel
marks variant with soft-filter `HighOccurrenceFrq` variant unless: Cancer_Somatic_Panel_Frq <= 0.3 (or) Cancer_Somatic_Panel_Frq == "."
Whole Genome Sequencing (WGS)#
Calling and quality filtration#
This section focuses on calling and quality filters.
Sentieon’s TNscope#
BALSAMIC utilizes the TNscope algorithm for calling somatic SNVs and INDELS in WGS samples. The TNscope algorithm performs the somatic variant calling on the tumor-normal or the tumor-only samples.
TNscope filtering (Tumor_normal)#
Pre-call Filters
Apply TNscope trained MachineLearning Model: Sets MLrejected on variants with ML_PROB below 0.32.
- ::
ML model: SentieonTNscopeModel_GiAB_HighAF_LowFP-201711.05.model is applied
min_init_tumor_lod: Initial Log odds for the that the variant exists in the tumor.
min_init_tumor_lod = 1
min_tumor_lod: Minimum log odds in the final call of variant in the tumor.
min_tumor_lod = 8
min_init_normal_lod: Initial Log odds for the that the variant exists in the normal.
min_init_normal_lod = 0.5
min_normal_lod: Minimum log odds in the final call of variant in the normal.
min_normal_lod = 1
Post-call Quality Filters
Total Depth (DP): Refers to the overall read depth from all target samples supporting the variant call
DP(tumor) >= 10 (or) DP(normal) >= 10
Allelic Depth (AD): Total reads supporting the ALT allele in the tumor sample
AD(tumor) >= 3
SOR: Symmetric Odds Ratio of 2x2 contingency table to detect strand bias
SOR < 4
Allelic Frequency (AF): Fraction of the reads supporting the alternate allele
Minimum AF(tumor) >= 0.05
alt_allele_in_normal: Default filter set by TNscope was considered too stringent in filtering tumor in normal and is removed.
bcftools annotate -x FILTER/alt_allele_in_normal
Relative tumor AF in normal: Allows for maximum Tumor-In-Normal-Contamination of 30%.
hard filters variants if: AF(normal) / AF(tumor) > 0.3
TNscope filtering (tumor_only)#
Pre-call Filters
min_init_tumor_lod: Initial Log odds for the that the variant exists in the tumor.
min_init_tumor_lod = 1
min_tumor_lod: Minimum log odds in the final call of variant in the tumor.
min_tumor_lod = 8
The somatic variants in TNscope raw VCF file (SNV.somatic.<CASE_ID>.tnscope.all.vcf.gz) are filtered out for the genomic regions that are not reliable (eg: centromeric regions, non-chromosome contigs) to enhance the computation time. This WGS interval region file is collected from gatk_bundles gs://gatk-legacy-bundles/b37/wgs_calling_regions.v1.interval_list.
Post-call Quality Filters
Total Depth (DP): Refers to the overall read depth supporting the variant call
DP(tumor) >= 10
Allelic Depth (AD): Total reads supporting the ALT allele in the tumor sample
AD(tumor) > 3
Allelic Frequency (AF): Fraction of the reads supporting the alternate allele
Minimum AF(tumor) > 0.05
SUM(QSS)/SUM(AD) >= 10
Read Counts: Count of reads in a given (F1R2, F2R1) pair orientation supporting the alternate allele
ALT_F1R2 > 0, ALT_F2R1 > 0
SOR: Symmetric Odds Ratio of 2x2 contingency table to detect strand bias
SOR < 4
Post-call Observation database Filters#
Note
Since BALSAMIC <= v18.0.0 Post-call database filters are initially applied as soft-filters and can be whitelisted in a subsequent step, whereupon they will be PASS
This section contains population database frequency filters.
GNOMADAF_POPMAX: Maximum Allele Frequency across populations
GNOMADAF_popmax <= 0.001 (or) GNOMADAF_popmax == "."
Normalized base quality scores: The sum of base quality scores for each allele (QSS) is divided by the allelic depth of alt and ref alleles (AD)
SWEGENAF <= 0.01 (or) SWEGENAF == "."
Frq: Frequency of observation of the variants from normal Clinical samples
Frq <= 0.007 (or) Frq == "."
Rescue and final filtration of variants#
Since BALSAMIC <= v18.0.0 Post-call database filters are initially applied as soft-filters. After this step the variants have a chance to be rescued if they meet either of the following criteria:
The variant is marked with CLNSIG=Pathogenic and/or CLNSIG=Likely_pathogenic and/or contains the ONC field that partially matches “oncogenic”, “tier” or “uncertain” in the INFO-field coming from Clinvar.
The variant exists in the optionally supplied whitelist VCF-file. The file in BALSAMIC/analysis_metadata/marked_causative.vcf will be used by default, but can be replaced by the –rescue-list option.
If either of those criteria are met the filters will be moved to a new INFO field called RescueFilters and the variant will be set to PASS, in addition the reason for the variant rescue will be given in the new RescueStatus INFO-field.
Finally the soft-filters will be applied as only variants marked as PASS or any of the allowed soft-filters will remain in the final vcf file (SNV.somatic.<CASE_ID>.<merged/tnscope>.<research/clinical>.filtered.pass.vcf.gz).
Attention: BALSAMIC <= v8.2.10 uses GNOMAD_popmax <= 0.005. From Balsamic v9.0.0, this settings is changed to 0.02, to reduce the stringency. BALSAMIC >= v11.0.0 removes unmapped reads from the bam and cram files for all the workflows. BALSAMIC >= v13.0.0 keeps unmapped reads in bam and cram files for all the workflows. BALSAMIC >= v16.0.0 uses UMIs for duplicate removal bam in standard TGA workflows.